Abklärung der Linksventrikelhypertrophie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Abklärung der Linksventrikelhypertrophie (LVH) rückt in den Fokus. Es stehen diverse neue Therapiemöglichkeiten sowohl bei hypertropher Kardiomyopathie (HCM) bzw. hypertropher obstruktiver Kardiomyopathie (HOCM) als auch in der Transthyretin-Amyloidose (ATTR-Amyloidose) zur Verfügung.
Keypoints
-
Symptomatische Patient:innen mit LVH sollten weiter abgeklärt werden.
-
Die frühe Diagnose und Therapie bei HCM/HOCM, kardialer Amyloidose oder M. Fabry sind prognoseentscheidend.
-
Die Echokardiografie ist als erste Untersuchungsmodalität bei LVH essenziell.
-
Das Herz-MRT stellt den Goldstandard zur weiteren Abklärung jeglicher Kardiomyopathie dar.
Auf kaum einem Gebiet in der Kardiologie gab es in den letzten Jahren so viele neue Entwicklungen wie bei Erkrankungen, die mit einer Linksventrikelhypertrophie (LVH) assoziiert sind. Dies hat auch zu einer erhöhten Sensibilität für die Diagnose der LVH geführt. Gleichzeitig handelt es sich um einen der häufigsten echokardiografischen Befunde. Mit diesem Spannungsfeld setzt sich der folgende Artikel auseinander.
Die echokardiografische Inzidenz der LVH wird in der Literatur in gesunden Kollektiven mit ca. 10% angegeben.1 In Kohorten mit bekannter arterieller Hypertonie steigt dieser Wert auf ca. 36–41%.2 Im klinischen Alltag ist es meist nicht möglich, alle diese Patient:innen in Hinblick auf die Ätiologie der LVH zu untersuchen. In der Großzahl der Fälle liegt der Herzmuskelverdickung ein erhöhter linksventrikulärer Füllungsdruck zugrunde. Bei jüngeren Patient:innen ist die häufigste Ätiologie die arterielle Hypertonie. Im fortgeschrittenen Alter überwiegen die Herzklappenvitien, insbesondere die Aortenklappenstenose. Unabhängig von der Ätiologie stellt die LVH einen Risikofaktor für kardiovaskuläre Morbidität und Mortalität dar.3
Eine Indikation zur weiterführenden Abklärung sind eine damit assoziierte Symptomatik, wie Dyspnoe, Leistungsknick, Synkope, Schwindel u.Ä., und ein Screening bei positiver Familienanamnese. Weiters sollten Patient:innen mit Red Flags für infiltrative myokardiale Erkrankungen sowie solche mit Zufallsbefunden hochgradiger Hypertrophie (≥15mm) näher untersucht werden. Unter den selteneren Erkrankungen, die mit einer Herzmuskelverdickung einhergehen, finden sich auch rasch progrediente, maligne Erkrankungen wie die AL-Amyloidose oder Erkrankungen mit erhöhtem Risiko für plötzlichen Herztod wie die HCM bzw. HOCM. Eine korrekte und frühzeitige Diagnose kann für diese Patient:innen prognoseentscheidend sein.
Symptomatik der LVH
Die arterielle Hypertonie als häufigste Ursache der LVH verläuft häufig über lange Jahre asymptomatisch. Symptome wie Kopfschmerzen, Schwindel, Ohrensausen oder Hitzegefühl sind selten und unspezifisch. Eine ausführliche und gezielte Anamnese ist deshalb unerlässlich, insbesondere die Erstdiagnose, aktuelle antihypertensive Therapie sowie Blutdruckkontrolle in den letzten Jahren bzw. Jahrzehnten. Eine langjährige unkontrollierte und therapieresistente Hypertonie ist stark mit der Entwicklung einer LVH vergesellschaftet.4 Unabhängig vom Vorliegen einer arteriellen Hypertonie ist auch die Sportanamnese zu berücksichtigen. Hier ist jedoch zu beachten, dass nur 2–13% männlicher (Ausdauer-)Sportler eine LVH im Ausmaß von 12–15mm entwickeln, bei Athletinnen ist die Prävalenz noch geringer.5
Typische Symptome können je nach Erkrankung auftreten (Tab.1). Bei HCM bzw.HOCM sind vor allem Patient:innen mit Obstruktion (ab ca. 30mmHg) von Symptomen betroffen, etwa von Dyspnoe, Schwindel, Synkopen oder Palpitationen. Typischerweise ist auch die Familienanamnese auffällig, da oft mehrere Familienmitglieder betroffen sind. Aufgrund des erhöhten Arrhythmierisikos ist die HCM bzw. HOCM einer der häufigsten Gründe des plötzlichen Herztods bei jungen Menschen.6
Hinweis auf eine kardiale Amyloidose können, neben Luftnot und Leistungseinschränkung, bilaterale Karpaltunnelsyndrome, Bizepssehnenrupturen, Störungen im autonomen Nervensystem oder Polyneuropathie geben. In späteren Krankheitsstadien leiden die Patient:innen an einer progredienten restriktiven Kardiomyopathie (HFpEF) mit höhergradigen NYHA-Stadien und Ödemen. Das Risiko für bradykarde Herzrhythmusstörungen ist deutlich erhöht, gegebenenfalls ist eine Herzschrittmacherimplantation indiziert.
Vorhofflimmern tritt bei HCM/HOCM oder kardialer Amyloidose häufig auf und wird oft schlecht toleriert. Eine orale Antikoagulation ist bei diesen Patient:innen ungeachtet des CHA2DS2-VA-Scores indiziert. Bei kardialer Dekompensation sollte großzügig eine stationäre Behandlung erwogen werden.
Lysosomale Speichererkrankungen, wie M.Fabry, M.Pompe oder M.Danon, treten oft in deutlich früheren Lebensjahren auf als die zuvor genannten Erkrankungen und sind mit syndromalen weiteren Pathologien, wie Seh- oder Hörstörungen, Nierenfunktionsstörungen, Muskelschwäche oder mentaler Retardierung assoziiert. Gerade beim M.Fabry gibt es jedoch Betroffene, die erst im Erwachsenenalter symptomatisch werden.
Abklärung/Diagnose der LVH
Elektrokardiografie (EKG)
Einen Hinweis auf das Vorliegen eines verdickten Herzmuskels kann das EKG geben. Tiefe S-Ausschläge über V1/2 und überhöhte R-Zacken in den lateralen Ableitungen sind typisch. Hierüber können der Sokolow-Lyon-Index oder der Lewis-Index bestimmt werden. Ebenfalls sehr etabliert sind die Cornell-Kriterien oder die Bestimmung des Cornell-Produkts, in das auch die Dauer des QRS-Komplexes einfließt. Auch weitere Beurteilungskriterien, wie die Peguero-Lo-Presti-Kriterien, sind in der Literatur beschrieben, allerdings in der klinischen Routine weniger etabliert. Gemein ist allen EKG-Kriterien eine niedrige Sensitivität bei moderater Spezifität, weshalb ein solcher Hinweis immer auch eine Bildgebung nach sich ziehen sollte.7 Auch eine Achsenabweichung nach links und typische Veränderungen der ST-Strecken und T-Wellen in den präkordialen Ableitungen können hinweisgebend sein (Abb.1).
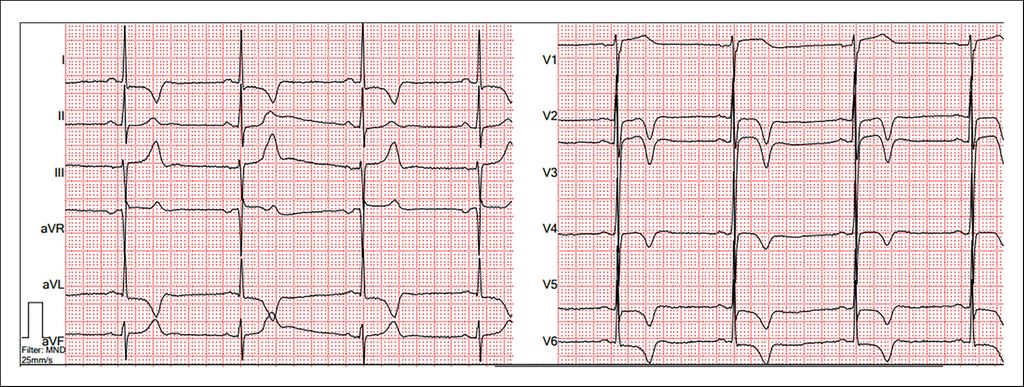
Abb. 1: EKG bei HCM/HOCM mit positivem Sokolow-Lyon-Index und sekundären Repolarisationsstörungen
Bei ATTR-Amyloidose fehlen diese typischen Kriterien häufig. Das EKG kann eine paradoxe Niedervoltage, ein Pseudoinfarktmuster mit Q-Zacken und eine träge R-Progression zeigen.
Echokardiografie
Zur echokardiografischen Beurteilung einer LVH wird üblicherweise der Durchmesser des intraventrikulären Septums und der Posterolateralwand herangezogen. Hier gelten bereits Werte >9mm bei Frauen und >10mm bei Männern als verdickt. Methodenbedingt werden die Wanddicken in der Echokardiografie jedoch häufig überschätzt (Abb.2). Definiert ist die LVH deshalb über die myokardiale Masse bezogen auf die Körperoberfläche (BSA; w: ≤95g/BSA; m: ≤115g/BSA), einen Parameter, der in der klinischen Praxis kaum verwendet wird.8
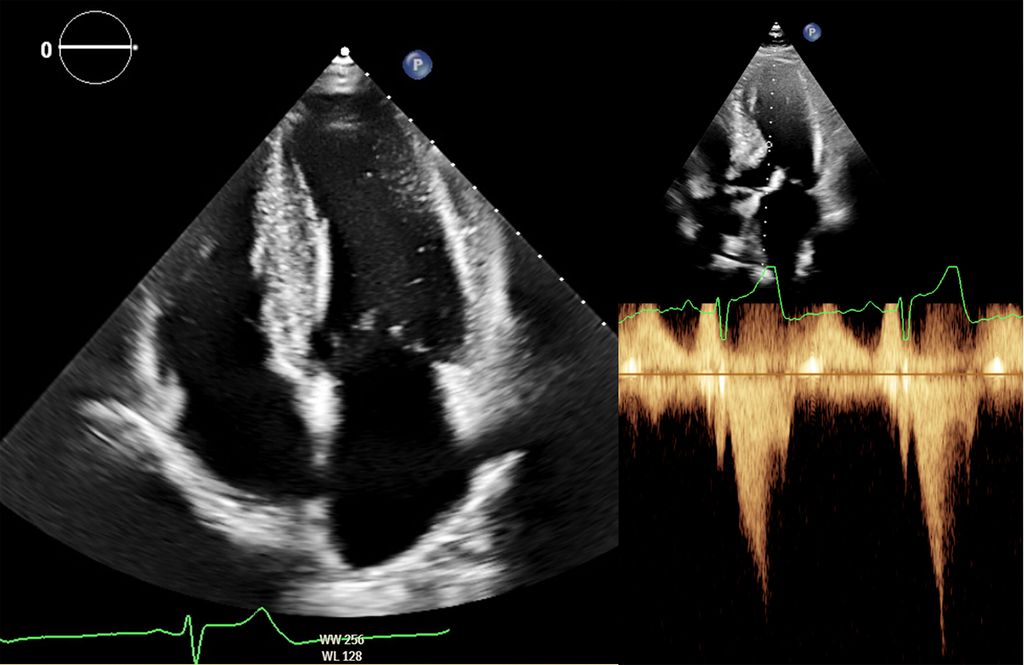
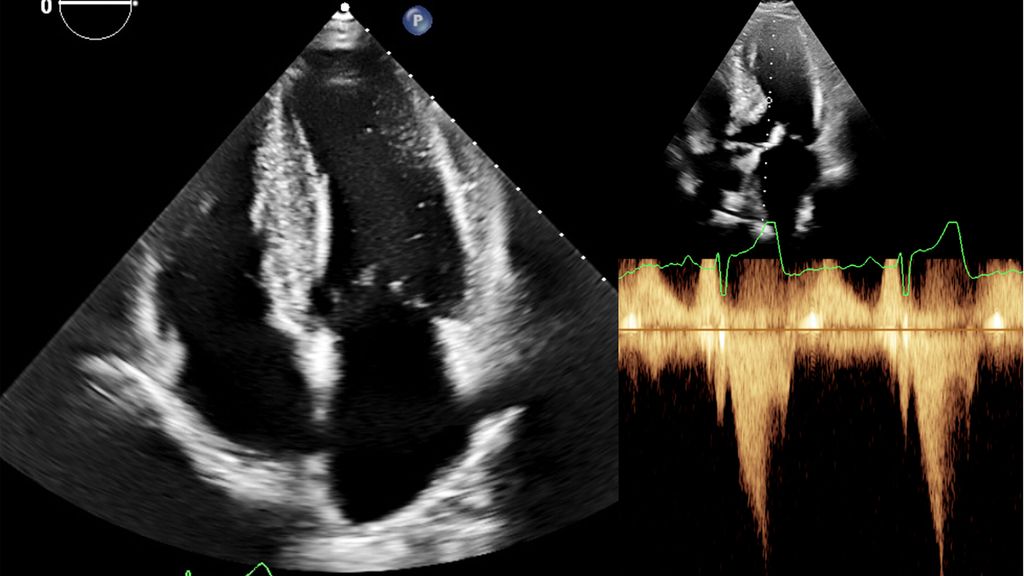
Abb. 2: Echokardiografie bei HOCM mit 4-Kammer-Blick (links) und Obstruktion im Ausflusstrakt (rechts)
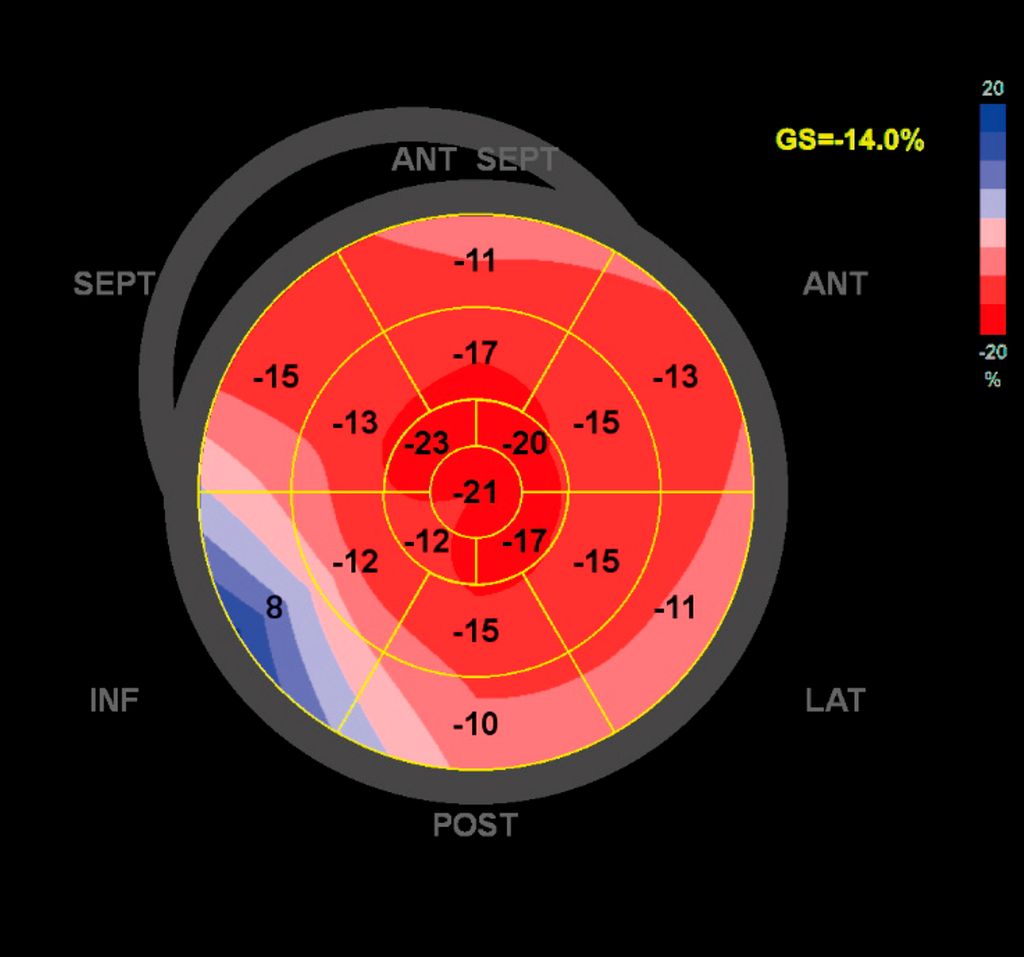
Abb. 3:Myokardiale Strainanalyse bei ATTR-Amyloidose
Dennoch ist die Echokardiografie aufgrund der niederschwelligen Verfügbarkeit als erste Untersuchungsmodalität und zur Verlaufsbeurteilung nicht wegzudenken. Mit der Ejektionsfraktion, der Vorhofgröße und der Beurteilung der diastolischen Funktion können prognostisch wichtige Parameter erhoben werden. Ein „apical sparing“ in der Analyse des myokardialen Strains oder der Aspekt eines „granular sparkling“ kann einen Verdacht auf kardiale Amyloidose begründen (Abb.3). Jede echokardiografische Untersuchung wegen des Verdachts auf LVH sollte außerdem eine Bestimmung des LVOT-Gradienten inkl. Valsalva-Manöver beinhalten (Abb.2).
Laboruntersuchungen/Elektrophorese
In Rahmen der Abklärung sollte eine Laboruntersuchung mit Blutbild, Nierenfunktionsparametern, Leberfunktionsparametern, Eisenstatus, Schilddrüsenwerten und NT-proBNP, hs-Troponin sowie Harnuntersuchung mit Proteinuriediagnostik erfolgen. Die Nierenfunktion und die Herzenzyme sind zur Stadienbeurteilung der zugrunde liegenden Erkrankung (z.B. bei ATTR-Amyloidose) unerlässlich.
Bei Verdacht auf kardiale Amyloidose muss zunächst mittels Elektrophorese die Bestimmung der freien Leichtketten inkl. Immunfixation im Serum und Harn erfolgen. Bei pathologischem Befund ist eine rasche hämatologische Vorstellung mit Verdacht auf AL-Amyloidose angezeigt.10 Auch verschiedene molekulargenetische Untersuchungen können im Verlauf der Abklärung der Linksventrikelhypertrophie indiziert sein. Hierzu gehören die Beurteilung der AGLA-Aktivität bei Verdacht auf M.Fabry sowie des TTR-Gens bei ATTR-Amyloidose oder die Bestimmung des CYP2C19-Metabolismus vor Therapie mit Mavacamten. Ein Genpanel oder ein „whole-exome sequencing“ ist bei Diagnose einer sarkomeren HCM/HOCM inklusive genetischer Beratung und Stammbaumanalyse indiziert. Weitere spezifische genetische Untersuchungen können bei sehr seltenen Ursachen der LVH indiziert sein, beispielsweise bei Glykogenosen, RASopathien oder mitochondrialen Myopathien.
Herz-Magnetresonanztomografie
Wenn der Verdacht auf eine zugrunde liegende strukturelle Herzerkrankung besteht, dann ist die kardiale Magnetresonanztomografie (MRT) der Goldstandard zur weiteren bildgebenden Abklärung. In der aktuellen Leitlinie der ESC hat die kardiale MRT-Untersuchung deshalb eine Klasse-I-Indikation im Rahmen der Erstabklärung sowie eine Klasse-IIa-Empfehlung beim Follow-up.11
Neben einer reproduzierbareren Beschreibung der kardialen Geometrie sowie einer exakten Messung der kardialen Funktion ist auch eine Gewebebeurteilung mittels T1- und T2-gewichteten Mappings zur differenzialdiagnostischen Beurteilung der zugrunde liegenden Kardiomyopathie möglich. Mit dem „late gadolinium enhancement“ (LGE) können außerdem Fibroseareale dargestellt werden und in die Risikostratifizierung einfließen. Hier konnte gezeigt werden, dass Patient:innen mit HCM/HOCM und >15% Fibrose des LV oder LVEF<50% ein erhöhtes Risiko für maligne Herzrhythmusstörungen bzw. plötzlichen Herztod (SCD) haben, weshalb eine primärprophylaktische Therapie mit ICD unabhängig von anderen klinischen Charakteristika erwogen werden kann.11
Über die Bestimmung des extrazellulären Volumens (ECV) kann außerdem der Verdacht auf eine Speichererkrankung erhärtet werden (Abb.4). Auch seltene Erkrankungen wie M.Fabry haben charakteristische Befundkonstellationen im kardialen MRT (Tab.1).12
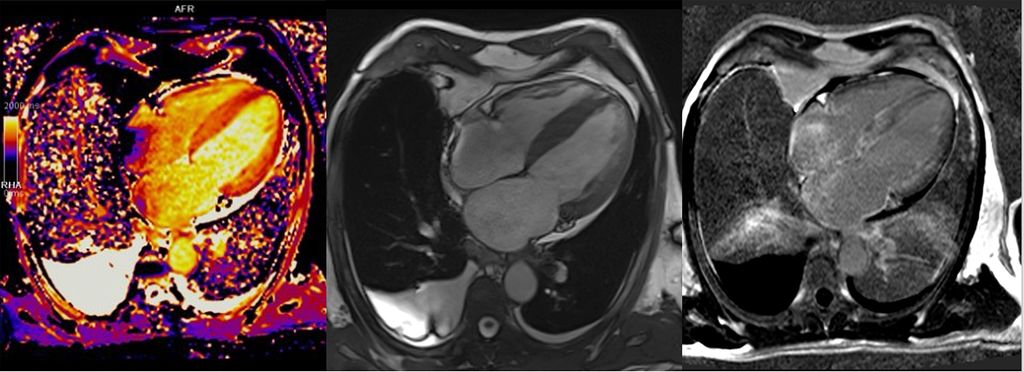
Abb. 4:Kardiale MRT bei ATTR-Amyloidose: erhöhtes T1-Mapping (links), 4-Kammer-Bick mit Hypertrophie (Mitte), diffuses subendokardiales LGE (rechts)

Tab. 1: Ursachen für LVH, häufige Symptome und typische MRT-Befundkonstellationen (modifiziert nach Martinez-Naharro et al. 2020)12
Knochenszintigrafie: PYP/DPD-Scan
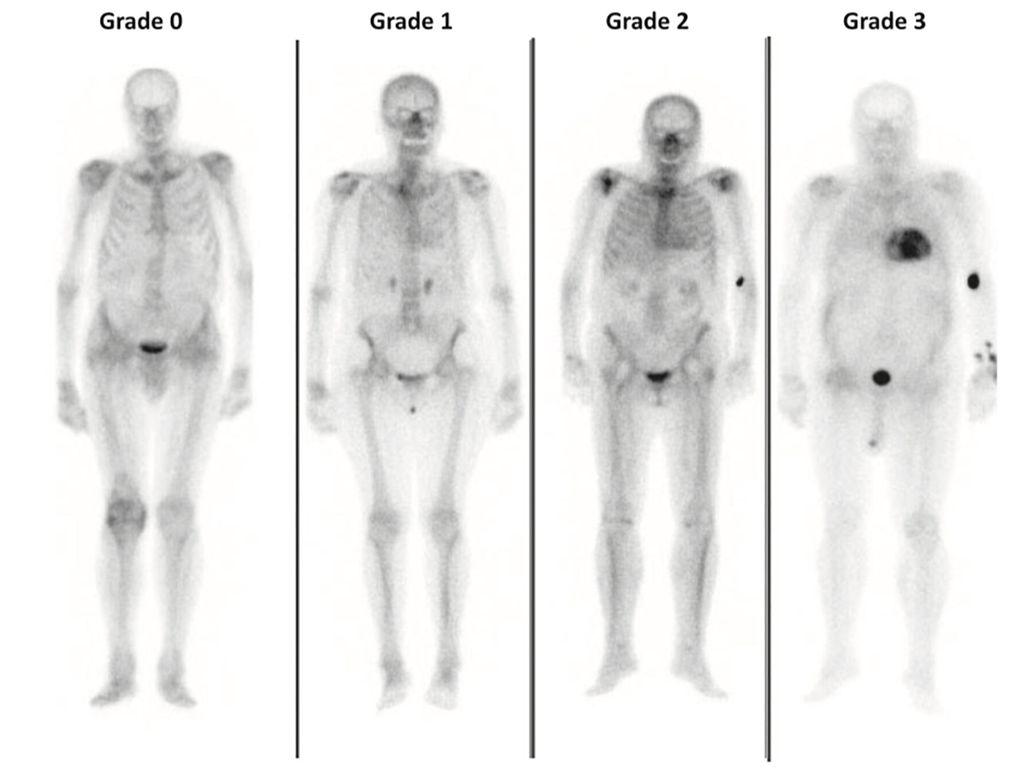
Abb. 5:Knochenszintigrafie mit Perugini-Score 0 (links) bis 3 (rechts; aus Garcia-Pavia P et al. 2021)9
Bei Verdacht auf kardiale Speichererkrankung/Amyloidose sollte eine Knochenszintigrafie mit Bestimmung des Perugini-Scores erfolgen (Abb.5).9 Dieser semiquantitative Score ermöglicht (bei Grad 2/3) die Diagnose einer ATTR-Amyloidose ohne histologische Gewebebeurteilung oder kann diese ausschließen (Grad 0). Im Falle eines Perugini Grad 1 ist eine weitere Abklärung über ein Zentrum indiziert. Zu beachten ist, dass die Knochenszintigrafie nicht verlässlich zwischen ATTR- und AL-Amyloidose unterscheiden kann. Aus diesem Grund müssen immer auch eine Elektrophorese und Immunfixation in Serum und Harn durchgeführt werden.13
Spezialambulanz für LVH
Wenn sich die LVH im MRT bestätigt, sollte die weitere Abklärung und Therapie zusammen mit einer Spezialambulanz für Linksventrikelhypertrophie erfolgen. Hier können nicht nur weiterführende Untersuchungen wie die Myokardbiopsie durchgeführt werden. Der Beginn und die Kontrolle der spezifischen Therapien müssen durch ein Zentrum erfolgen. Bei Verdacht auf eine genetische Ursache sollte in weiterer Folge auch eine genetische Testung inklusive Stammbaumanalyse und genetischer Beratung veranlasst werden.
Hypertrophe Kardiomyopathie
Die hypertrophe Kardiomyopathie (HCM/HOCM) ist mit einer Inzidenz von 0,2% die häufigste genetische Kardiomyopathie. Sie ist definiert über einen Linksventrikeldurchmesser ≥15mm in einem beliebigen LV-Segment, der nicht durch erhöhte Füllungsdrücke erklärbar ist. Geringere Wanddicken (13–14mm) sind insbesondere bei positiver Familienanamnese zu berücksichtigen.11 Bei der obstruktiven Form leiden die Betroffenen häufig unter Leistungseinschränkung, Belastungsdyspnoe und Schwindel. Ein Ausflussgradient (LVOTO) ≥50mmHg in Ruhe oder unter Belastung (Valsalva-Manöver) gilt als Therapieindikation für Betablocker oder nDHP-Kalziumkanalblocker. Bei anhaltender Symptomatik gibt es seit 2024 mit Mavacamten, dem ersten Wirkstoff aus der Gruppe der Myosininhibitoren, eine effektive und gut verträgliche medikamentöse Therapieoption.14 In der VALOR-HCM-Studie konnte außerdem gezeigt werden, dass unter Mavacamten deutlich seltener eine Indikation für septale Reduktionstherapien (Alkoholablation/Myektomie) besteht.15
Bei allen Patient:innen mit HCM/HOCM sollte eine regelmäßige Risikostratifizierung erfolgen. Wir empfehlen, zumindest jährlich ein Holter-EKG durchführen zu lassen, da nichtanhaltende ventrikuläre Tachykardien ein unabhängiger Risikofaktor für den plötzlichen Herztod sind. Alle 3–5 Jahre sollte das Risikoprofil mit dem HCM-Risk-Score berechnet und es sollten das ausgeprägte Fibroseareal im MRT (≥15%) und die reduzierte LVEF (≤50%) berücksichtigt werden.11 Bei HOCM sind Volumenmangelzustände (Dehydratation, Alkoholexzess) sowie Nitrate und PDE5-Inhibitoren zu vermeiden.
Kardiale Amyloidosen
Amyloidose ist ein Überbegriff für Erkrankungen, die auf der Einlagerung von Proteinfilamenten in den Extrazellulärraum basieren. Hierdurch kommt es zu einer Funktionseinschränkung des betreffenden Gewebes. Im Fall der ATTR-Amyloidose ist eine Mutation im TTR-Gen oder ein Alterungsprozess für die Instabilität des Transthyretins verantwortlich. Betroffene leiden unter progredienten Symptomen einer restriktiven Herzinsuffizienz/HFpEF. Die medikamentöse Therapie sollte (neben der spezifischen Therapie) einen SGLT2-Inhibitor, einen Mineralokortikoidantagonisten und, bei Ödembildung, ein Schleifendiuretikum beinhalten. Bei Verschlechterung der Linksventrikelfunktion ist eine neurohumorale Therapie prinzipiell indiziert, wird jedoch in diesem Kollektiv nicht von allen Patient:innen gut toleriert.9
Um den weiteren Progress der Erkrankung zu verlangsamen, stehen mittlerweile Tafamidis und seit 2025 auch Acoramidis aus der Gruppe der TTR-Stabilisatoren als erstattungsfähige Therapien zur Verfügung.16,17 2025 wurde außerdem mit Vutrisiran ein Vertreter aus der Gruppe der „gene silencer“ durch die EMA zugelassen.18
Fazit
Die Linksventrikelhypertrophie ist ein sehr häufiger Befund bei internistischen Patient:innen. Symptomatik und Red Flags können auf eine spezifische zugrunde liegende Erkrankung hinweisen. Die kardiale Bildgebung mittels Echokardiografie und MRT dient der Objektivierung des Verdachts und der Differenzialdiagnose. Meist gelingt eine Diagnosestellung nichtinvasiv in Zusammenschau mit Laboruntersuchungen, molekulargenetischen Bestimmungen und Knochenszintigrafie. Für HCM/HOCM, ATTR-Amyloidose und M.Fabry stehen spezifische Therapien zur Verfügung, die, in Kombination mit adäquater Risikostratifizierung und regelmäßigen Kontrollen, die Lebensqualität und Prognose der Patient:innen entscheidend verbessern können.
Literatur:
1 Schirmer H et al.: Prevalence of left ventricular hypertrophy in a general population. The Tromsø study. Eur Heart J 1999; 20(6): 429-38 2 Cuspidi C et al.: Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens 2012; 26(6): 343-9 3 Bombelli M et al.: Impact of the increase in left ventricular mass on the risk of long-term cardiovascular mortality: a prospective cohort study. Hypertension 2023; 80(6): 1321-30 4 Izzo R et al.: Development of left ventricular hypertrophy in treated hypertensive outpatients: the campania salute network. Hypertension 2017; 69(1): 136-42 5 Pelliccia A et al.: 2020 ESC guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J 2021; 42(1): 17-96 6 Link MS: Sudden cardiac death in the young: epidemiology and overview. Congenit Heart Dis 2017; 12(5): 597-9 7 Noubiap JJ et al.: A meta-analytic evaluation of the diagnostic accuracy of the electrocardiographic Peguero-Lo Presti criterion for left ventricular hypertrophy. J Clin Hypertens (Greenwich) 2020; 22(7): 1145-53 8 Lang RM et al.: Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015; 28(1): 1-39 9 Garcia-Pavia P et al.: Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2021; 42(16): 1554-68 10 Writing Committee et al.: 2023 ACC Expert Consensus Decision Pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol 2023; 81(11): 1076-126 11 Arbelo E et al.: 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J 2023; 44(37): 3503-626 12 Martinez-Naharro A et al.: Diagnostic imaging of cardiac amyloidosis. Nat Rev Cardiol 2020; 17(7): 413-26 13 Gillmore JD et al.: Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133(24): 2404-12 14 Olivotto I et al.: Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020; 396: 759-69 15 Desai MY et al.: Mavacamten in patients with hypertrophic cardiomyopathy referred for septal reduction: week 56 results from the VALOR-HCM randomized clinical trial. JAMA Cardiol 2023; 8(10): 968-77 16 Maurer MS et al.: Tafamidis treatment for patients with Transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379(11): 1007-16 17 Gillmore JD et al.: Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med 2024; 390(2): 132-42 18 Fontana M et al.: Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J 2024; 392(1): 33-44
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
Adhärenz als Schlüsselfaktor für eine erfolgreiche Behandlung
Ein ausbleibender oder unzureichender Behandlungserfolg bedeutet nicht zwingend, dass die Therapie wirkungslos ist. Manchmal liegt es auch daran, dass die Medikation nicht eingenommen, ...